
Messages clés
- Un état de mal épileptique est une crise anormalement prolongée (> t1), dont la probabilité de résolution spontanée est faible.
- Sa physiopathologie passe par un déséquilibre entre excitation (augmentée) et inhibition (diminuée).
- Un état de mal épileptique qui se prolonge expose à un risque de lésions cérébrales (> t2) et de complications extraneurologiques.
- Les traitements doivent être utilisés lorsqu’ils sont indiqués (t1 dépassé) et leur retard d’administration peut exposer à une pharmacorésistance.
- Un état de mal épileptique prolongé peut nécessiter des traitements spécifiques : sédation (action GABAergique) et traitements modulant l’action des récepteurs AMPA et NMDA (pérampanel, kétamine).
Introduction
Selon Henri Gastaut en 1967, l’état de mal épileptique (EME) se caractérise par « une crise épileptique qui persiste suffisamment longtemps ou qui se répète à des intervalles suffisamment brefs pour créer une condition épileptique stable et durable ». Au-delà d’une certaine durée, cet état épileptique stable et prolongé entraîne des conséquences cérébrales irréversibles. La compréhension de la physiopathologie de l’EME est utile pour appréhender au mieux la prise en charge thérapeutique de l’EME et limiter au maximum les conséquences cérébrales ainsi que la mortalité.
Mécanismes physiopathologiques des crises et de l’EME
Dans une synapse excitatrice normale, le neurone présynaptique libère du glutamate qui agit sur les récepteurs post-synaptiques NMDA (acide N-méthyl-D-aspartique) et AMPA (classe de récepteurs sensibles à l’acide amino-hydroxy-méthyl-isoxazole-propionique), tandis que les interneurones GABAergiques (acide gamma-aminobutyrique de type A) assurent un rétrocontrôle inhibiteur. Les cellules gliales participent également à l’homéostasie synaptique en régulant les neurotransmetteurs extracellulaires et l’équilibre ionique. Les hypothèses physiopathologiques dans l’EME ont principalement été étudiées dans des modèles animaux.
La physiopathologie des crises d’épilepsie repose principalement sur un déséquilibre entre l’excitation et l’inhibition neuronale [1-3]. La grande majorité des crises d’épilepsie cessent spontanément grâce à plusieurs mécanismes biologiques, notamment la déplétion des neurotransmetteurs, la diminution de l’ATP, les modifications ioniques ou encore l’augmentation du GABA [1].
La transition entre une crise d’épilepsie qui cède spontanément et un état de mal épileptique s’explique essentiellement par une baisse des mécanismes d’inhibition neuronale, ce qui favorise un état d’excitation et une absence de retour à un état normal. Dans ce processus, les neurotransmetteurs excitateurs comme le glutamate, et les inhibiteurs comme le GABA jouent un rôle central (Fig. 1).
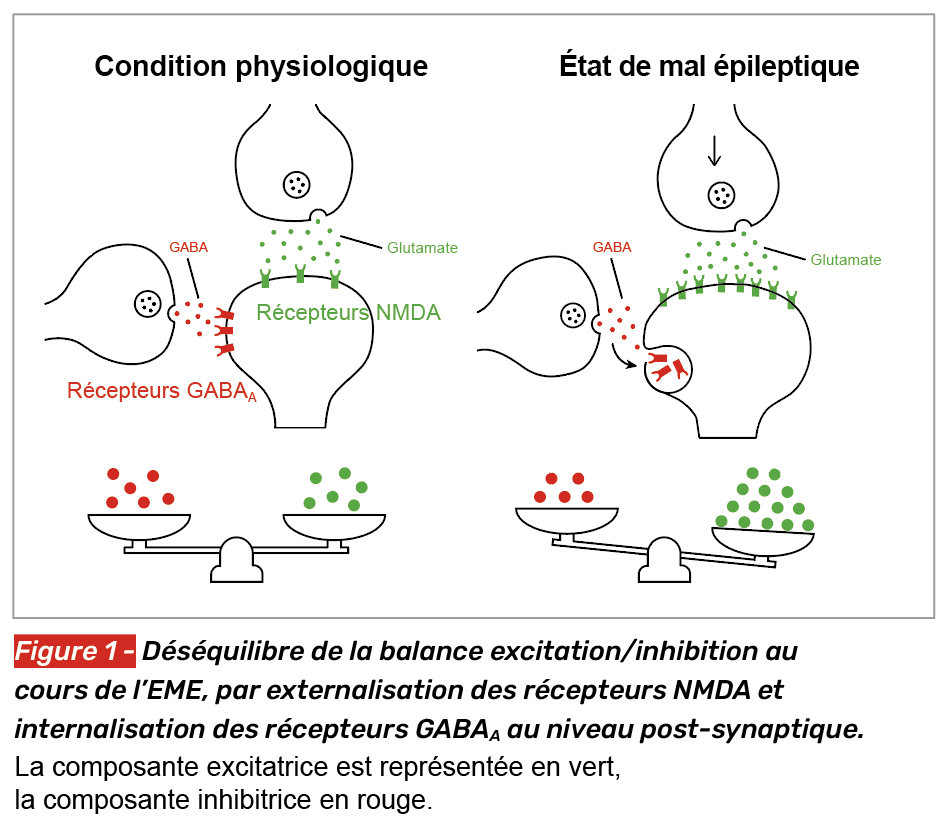
Baisse de l’inhibition
Dans les premiers temps de l’EME, les mécanismes inhibiteurs, principalement via le GABA, perdent de leur efficacité par une internalisation des récepteurs GABAA en post-synaptique, ce qui contribue à la baisse d’efficacité des benzodiazépines au cours de l’EME [1, 3-7].
Excitation neuronale excessive
Au cours de l’EME, les récepteurs glutamatergiques NMDA sont externalisés dans la membrane post-synaptique, ce qui favorise l’excitation neuronale [1, 3]. Par ailleurs, des modifications des récepteurs AMPA surviennent avec une perte de la sous-unité GluA2, rendant ces récepteurs perméables au calcium. Cette augmentation de l’entrée calcique favorise l’excitotoxicité neuronale, le stress oxydatif, la dysfonction mitochondriale et la mort cellulaire. Ce mécanisme constitue une cible thérapeutique de choix dans l’EME prolongé : les antagonistes des récepteurs NMDA comme la kétamine par voie intraveineuse, ou les antagonistes des récepteurs AMPA comme le pérampanel par voie entérale, pourraient être particulièrement indiqués à ce stade, où l’excitation glutamatergique post-synaptique prédomine et où l’efficacité des benzodiazépines pourrait être compromise.
Ce déséquilibre entre baisse de l’inhibition et augmentation de l’excitation s’aggrave au cours de l’EME, ce qui favorise l’auto-entretien de l’EME et une possible pharmacorésistance.
Lésions neuronales
Les lésions neuronales peuvent être liées :
• à l’EME lui-même, car il peut provoquer une mort neuronale via plusieurs mécanismes : accumulation intracellulaire excessive de calcium liée à l’hyperactivation des récepteurs NMDA, entraînant l’activation d’enzymes toxiques, un stress oxydatif, une dysfonction mitochondriale avec diminution de production d’ATP, une altération des membranes cellulaires et des pompes ioniques Na+/K+ ATPase. Ces mécanismes favorisent l’excitotoxicité neuronale et conduisent à l’apoptose ; s’y associent également une hypoxie, une toxicité du glutamate et une acidose lactique. Certaines structures comme l’hippocampe sont particulièrement vulnérables à cette excitotoxicité calcique, pouvant conduire à une sclérose hippocampique, des troubles mnésiques et des séquelles cognitives persistantes ;
• aux conséquences systémiques de l’état de mal (hypotension, hypoxie, hypoglycémie, acidose, etc.).
Évolution de l’EME : phases t1 et t2
Plus l’EME persiste, plus les conséquences cérébrales et systémiques peuvent devenir importantes.
Deux temps opérationnels ont été définis à partir du début de la crise, par la ligue internationale contre l’épilepsie : les temps t1 et t2 [10] (Fig. 2).
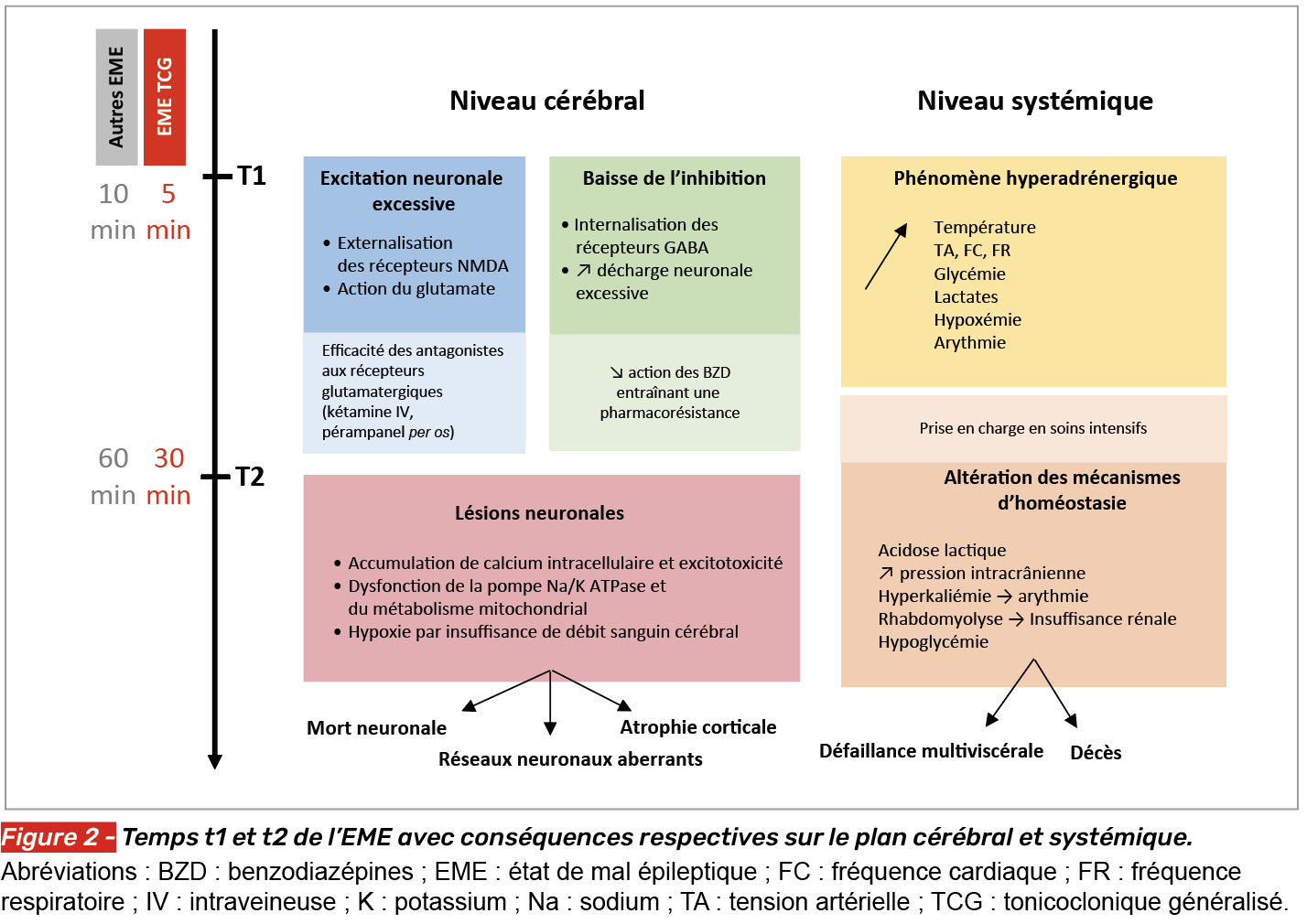
Temps t1 (phase aiguë, au-delà de 5 min pour l’EME généralisé tonico-clonique, 10 min pour les autres EME)
C’est le temps au-delà duquel une crise d’épilepsie est considérée comme un EME, du fait d’une probabilité de résolution spontanée faible. Dans les premières minutes, l’EME est marqué par une diminution des phénomènes inhibiteurs (internalisation des récepteurs GABAA) et augmentation des phénomènes excitateurs (externalisation des récepteurs NMDA). Sur le plan systémique, notamment pour l’EME généralisé tonico-clonique, il se produit un phénomène adrénergique via la surproduction de catécholamines associant une hyperthermie, une hypercapnie, une hypoxémie, une augmentation de la fréquence cardiaque, une augmentation de la pression artérielle et une hyperglycémie. Une acidose lactique apparaît, en relation avec les contractions musculaires intenses en situation d’anaérobie [2].
Temps t2 (phase prolongée, au-delà de 30 min pour l’EME généralisé tonico-clonique, > 60 min pour les autres EME)
C’est le temps au-delà duquel l’EME peut entraîner des lésions neuronales irréversibles, une mort neuronale et sur le long terme une atrophie corticale, des réseaux neuronaux aberrants et des conséquences cognitives [2, 3]. Certaines structures, notamment l’hippocampe, sont particulièrement vulnérables. Ces lésions et remaniements neuronaux peuvent favoriser une épileptogenèse secondaire avec constitution de réseaux hyperexcitables (conduisant au risque de maladie épileptique après un état de mal de novo). Sur le plan systémique, notamment pour l’EME généralisé tonico-clonique, l’hyperkaliémie liée à l’activation musculaire peut entraîner des arythmies et la rhabdomyolyse peut conduire à une insuffisance rénale. Par la suite surviennent une hypoglycémie, une acidose lactique marquée et une augmentation de la pression intracrânienne [3]. Tous ces phénomènes pouvant conduire à une défaillance multiviscérale et au décès.
Conclusion
Le passage d’une crise à un état de mal épileptique est sous-tendu par un processus physiopathologique complexe qui repose sur un dépassement des mécanismes protecteurs inhibiteurs qui font habituellement cesser une crise, puis par un déséquilibre de la balance inhibition/excitation au profit de l’excitation qui s’aggrave au cours du temps. Une bonne connaissance de la physiopathologie de l’EME est essentielle pour la prise en charge thérapeutique, en particulier pour le choix du traitement antiépileptique et du moment d’administration, afin de limiter les conséquences cérébrales et systémiques de l’EME.

Correspondance
s.george@chru-nancy.fr
j.jonas@chru-nancy.fr
Les auteurs déclarent ne pas avoir de lien d’intérêt.
Bibliographie
1. Walker MC. Pathophysiology of status epilepticus. Neuropharmacology 2016 ; 116 : 18-26.
2. Sánchez Fernández I, Goodkin HP, Scott RC. Pathophysiology of convulsive status epilepticus. Seizure 2019 ; 68 : 16–21.
3. Engrand N, Crespel A. Bases physiopathologiques des états de mal épileptiques. Rev Neurol 2009 ; 165 : 315–9.
4. Chen JWY, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand 2007 ; 115 : 7-15.
5. Tyvaert L. État de mal épileptique. Prat Neurol 2017 ; 8 : 70–9.
6. Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci 2005 ; 25 : 5511–20.
7. Naylor DE, Liu H, Wasterlain CG. Traffi cking of GABA(A) receptors, loss of inhibition,and a mechanism for pharmacoresistance in status epilepticus. J Neurosciences, 2005 ; 25 : 7724–33.
8. Hervé Outin, Gueye P, Alvarez V et al. Prise en charge des états de mal épileptiques en préhospitalier, en structure d’urgence et en réanimation dans les 48 premières heures. Recommandations formalisées d’experts SRLF – SFMU ; 2018.
9. Redecker J, Wittstock M, Benecke R, Rösche J. Efficacy of perampanel in refractory nonconvulsive status epilepticus and simple partial status epilepticus. Epilepsy Behav 2015 ; 45 : 176–9.
10. Trinka E, Cock H, Hesdorffer D et al. A defi nition and classifi cation of status epilepticus – Report of the ILAE Task Force on Classifi cation of Status Epilepticus. Epilepsia 2015 ; 56 : 1515-23.