
L’ataxie de Friedreich (FA) représente la plus fréquente des ataxies autosomiques récessives, avec une prévalence estimée entre 1/30 000 et 1/50 000 dans les populations caucasiennes. En Europe, environ 25 000 patients sont concernés, avec une fréquence de portage hétérozygote variant de 1/85 à 1/100 [1].
La FA dépasse le cadre d’une simple ataxie : elle s’accompagne de manifestations sensitives, pyramidales, cardiaques, endocriniennes, orthopédiques, et autres traduisant une atteinte multi-systémique. L’évolution conduit progressivement à une perte d’autonomie et à une altération marquée de la qualité de vie.
Bien qu’aucun traitement curatif ne soit disponible à ce jour, l’arrivée de l’omavéloxolone marque une avancée majeure et ouvre la voie à de nouvelles approches thérapeutiques ciblant les mécanismes mitochondriaux et transcriptionnels.
Bases moléculaires
La FA est causée par une expansion anormale du triplet GAA dans le premier intron du gène FXN, situé sur le chromosome 9q13 [2] :
• 96 à 98 % des patients présentent une expansion bi-allélique (600-900 répétitions en moyenne, extrêmes : 70-1 700) ;
• 2 à 4 % présentent une expansion mono-allélique associée à une mutation ponctuelle sur l’autre allèle, pouvant constituer un piège diagnostique [3].
Le gène FXN code la frataxine, protéine mitochondriale essentielle à l’assemblage et au transport des clusters fer-soufre (Fe-S). L’expansion du triplet GAA entraîne une méthylation anormale et ainsi à la formation d’hétérochromatine, aboutissant à un silence transcriptionnel et donc à un déficit en frataxine (Fig. 1).
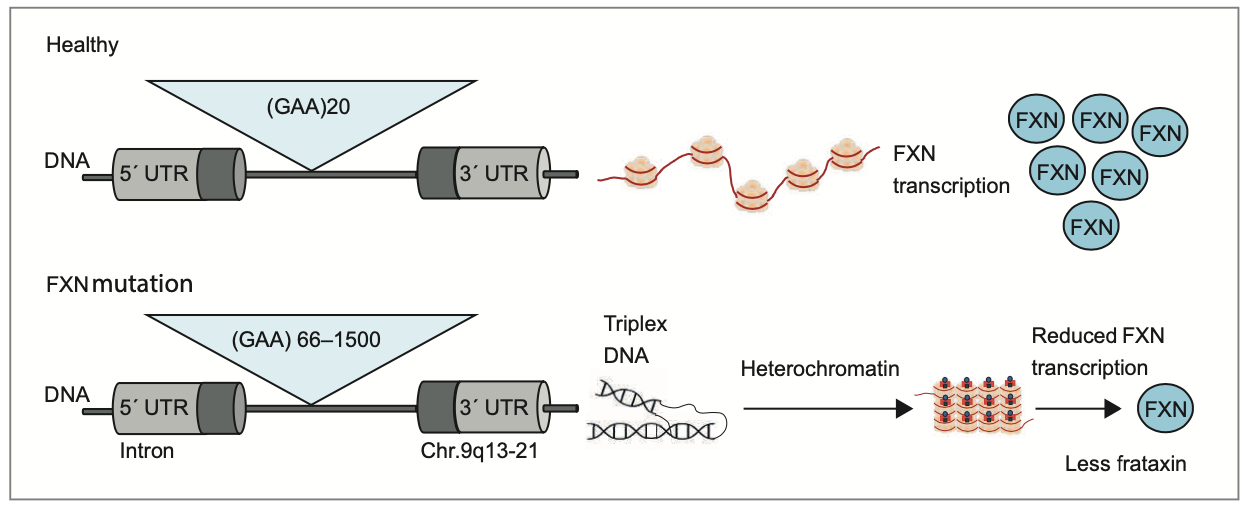
Figure 1 – Mécanisme physiopathologique impliqué dans l’ataxie de Friedreich [4].
Physiopathologie
La frataxine, localisée dans la membrane mitochondriale interne, intervient dans l’assemblage et le transport des clusters Fe-S, indispensables au fonctionnement de la chaîne respiratoire, à la régulation du fer et à la synthèse de l’hème [5].
Le déficit en frataxine provoque (Fig. 2) :
• une altération des enzymes Fe-S,
• une accumulation de fer mitochondrial (surtout cardiaque et cérébelleuse),
• un stress oxydatif majeur responsable de peroxydation lipidique et de déplétion en glutathion.
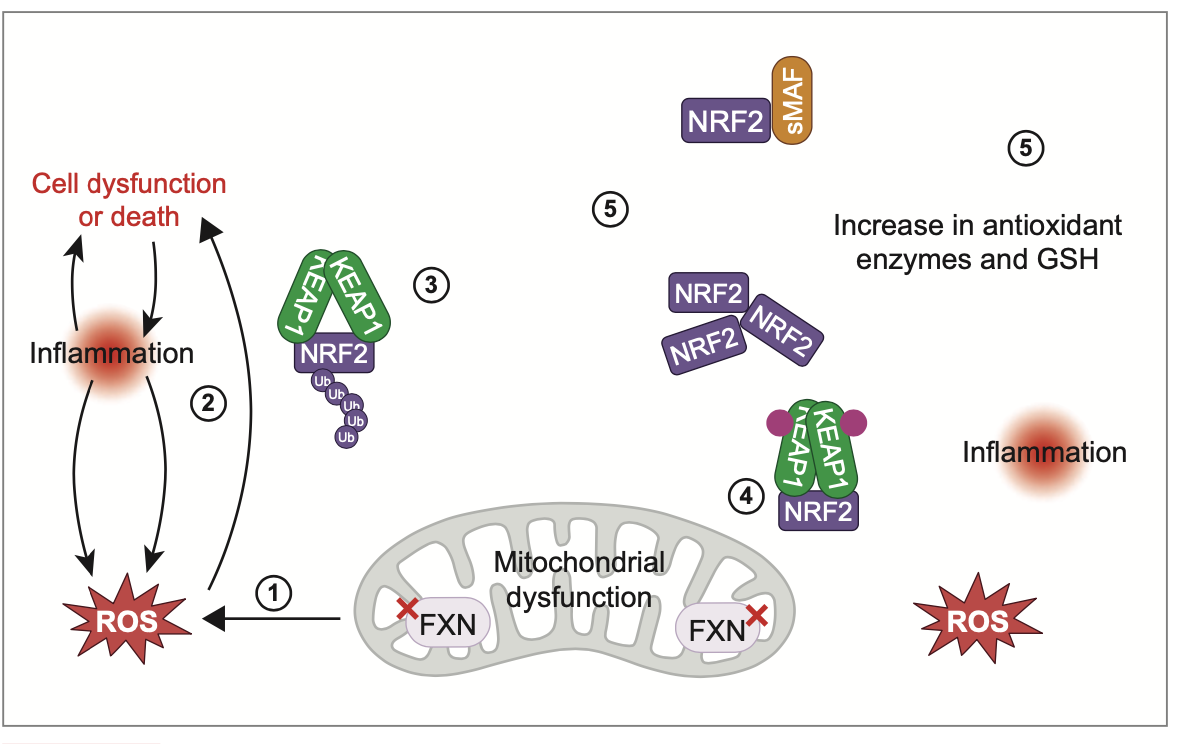
Figure 2 – Conséquences du déficit en frataxine dans la mitochondrie [6].
Ces anomalies aboutissent à une dégénérescence neuronale et myocardique progressive, faisant de la FA une maladie mitochondriale systémique.
Phénotypes et formes cliniques
On peut distinguer deux grandes formes cliniques [4].
Forme classique
Le début survient avant 25 ans, le plus souvent entre 10 et 15 ans, et l’âge de début est corrélé à la taille de l’expansion GAA [7]. Les premiers symptômes incluent l’instabilité de la marche (77 %), la scoliose (23 %) et les chutes répétées [7].
Elle associe une ataxie mixte, proprioceptive et cérébelleuse, à des complications extra-neurologiques fréquentes.
Manifestations neurologiques
Les signes inauguraux consistent en des troubles de la marche et de l’équilibre, avec démarche instable, chutes fréquentes et difficultés à courir.
L’examen neurologique permet de retrouver :
• une ataxie cérébelleuse : axiale, des membres et du tronc, associée à une dysarthrie ;
• une atteinte proprioceptive : secondaire à une atteinte médullaire (cordonale postérieure) et périphérique sensitive ;
• une abolition des réflexes ostéotendineux (ROT) ;
• un syndrome pyramidal : spasticité, signe de Babinski.
L’association caractéristique est celle d’une ataxie mixte (cérébelleuse et proprioceptive), spastique avec abolition des ROT apparaissant dans l’enfance ou l’adolescence.
De plus, les patients peuvent présenter de nombreux autres signes neurologiques, notamment une dysphagie, une neuropathie, une spasticité, des troubles dépressifs, des troubles du sommeil, un syndrome des jambes sans repos, des troubles urinaires ainsi que des douleurs [8].
Manifestations extra-neurologiques
Les manifestations extra-neurologiques de FA sont fréquentes et contribuent largement à la morbidité de la maladie. Elles concernent principalement les systèmes cardiaque, ostéoarticulaire, neurosensoriel et endocrinien [8].
Atteintes ostéoarticulaires (Fig. 3)
• La scoliose constitue la manifestation non neurologique la plus fréquente, observée chez 63 à 90 % des patients. Elle apparaît souvent précocement, parfois avant les premiers signes neurologiques. Elle résulte principalement de la faiblesse neuromusculaire et se caractérise typiquement par une courbure thoracique gauche et une hypercyphose. Les formes sévères nécessitent fréquemment une arthrodèse.
• Les déformations des pieds sont également fréquentes (≈ 60 %), dominées par le pied creux, lié à un déséquilibre des forces musculaires.
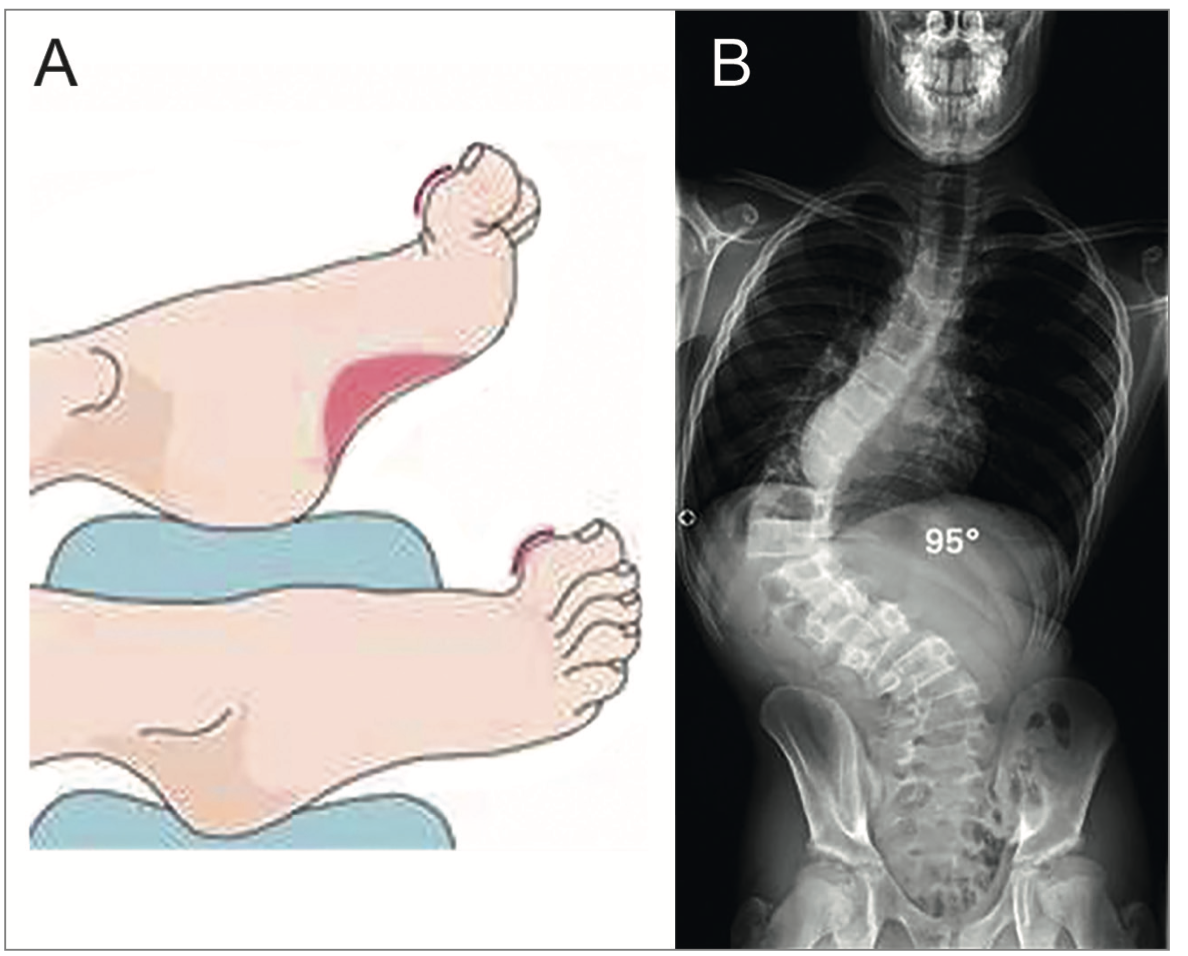
Figure 3 – Manifestations articulaires. A : pied creux ; B : scoliose [9].
Atteinte cardiaque
Présente chez 40 à 85 % des patients, elle représente la principale cause de mortalité.
• La cardiomyopathie hypertrophique concentrique est la forme la plus caractéristique, sans signe obstructif. Cette hypertrophie cardiaque est souvent associée à des ondes T négatives en inféro-latéral (en raison de l’hypertrophie). Disposer d’un ECG de référence permet de faire le diagnostic différentiel avec une souffrance myocardique d’origine ischémique sous-épicardique.
• Des troubles du rythme (fibrillation auriculaire, flutter, anomalies de conduction) et une insuffisance cardiaque surviennent dans environ 20 % des cas. L’évolution se fait progressivement, d’anomalies électriques précoces à une hypertrophie myocardique puis à une fibrose diffuse conduisant à la dysfonction cardiaque.
Atteinte neurosensorielle
• Elle est souvent visuelle, liée à une neuropathie optique (50-100 %), responsable d’une baisse de l’acuité visuelle, notamment à faible contraste (Fig. 4).
• Une atteinte auditive est également fréquente (90-100 %), mais souvent méconnue. Elle se manifeste par une dyssynchronie centrale et une difficulté de compréhension dans le bruit, malgré des audiogrammes parfois normaux.
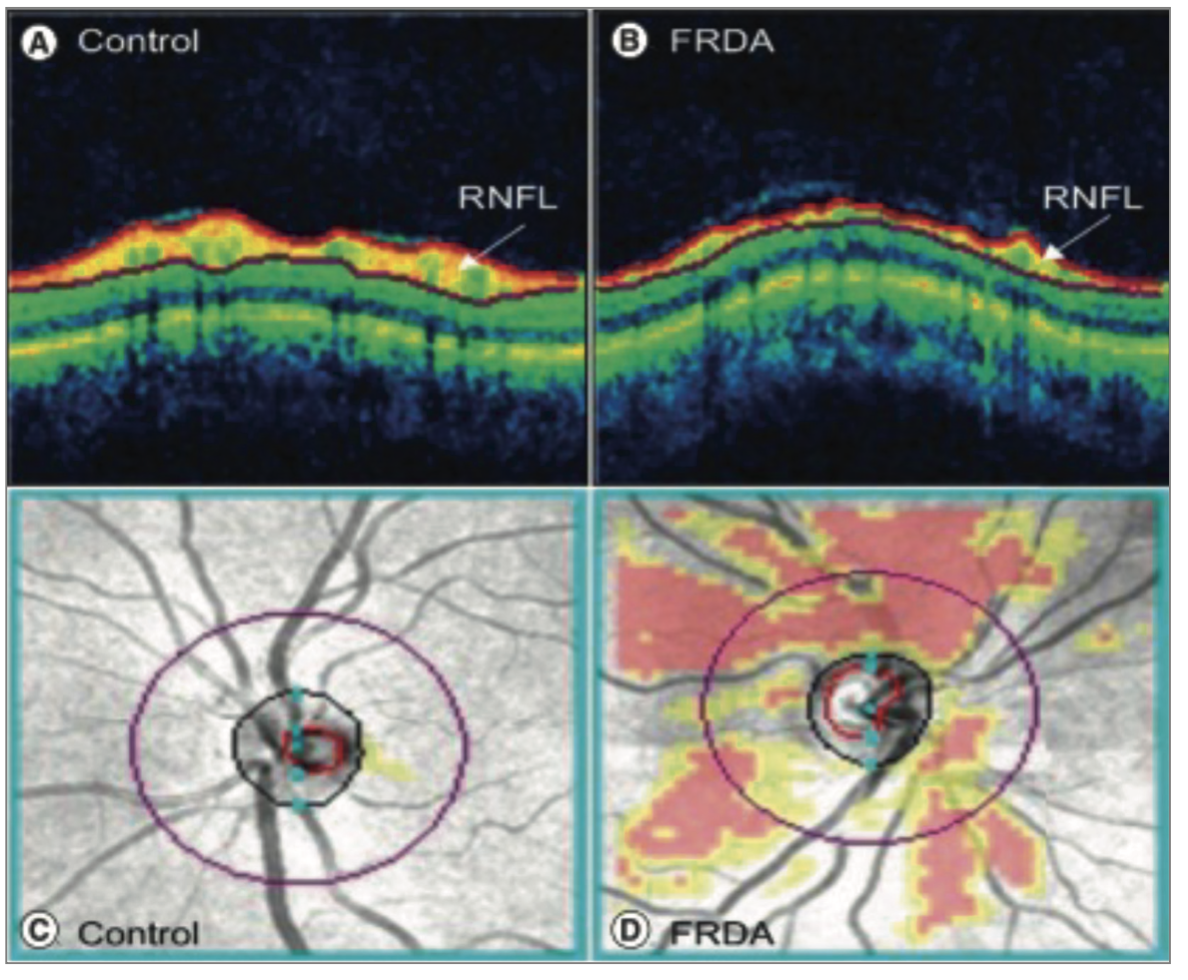
Figure 4 – Atrophie optique [10].
Atteinte endocrinienne
• Le risque de diabète est augmenté par rapport à la population générale, bien que sa prévalence demeure modérée : environ 30 % des patients présentent une intolérance au glucose et 7 à 9 %, un diabète avéré [4].
• Une tendance au surpoids est fréquemment observée chez l’adulte, liée possiblement à un déficit en frataxine dans le tissu adipeux blanc, entraînant hypertrophie adipocytaire, hypovascularisation et inflammation locale.
Particularités des formes tardives
Les formes à début tardif de FA représentent environ 17 % des cas [4]. On distingue classiquement :
• la LOFA (Late-Onset Friedreich Ataxia), lorsque les premiers symptômes apparaissent après 25 ans ;
• la vLOFA (very Late-Onset Friedreich Ataxia), lorsque le début survient après 40 ans. Des formes exceptionnellement tardives, débutant après 60 ans, ont également été rapportées.
Sur le plan clinique
Ces formes tardives se distinguent par une évolution plus lente et des symptômes généralement plus modérés que dans les formes précoces. Les atteintes cardiaques et ostéoarticulaires y sont moins fréquentes, tandis que certains signes neurologiques diffèrent :
• les réflexes ostéotendineux peuvent être conservés,
• la spasticité est souvent plus marquée, pouvant simuler une paraplégie spastique,
• la dysarthrie et les manifestations extra-neurologiques (cardiomyo-pathie, diabète, déformations squelettiques) sont moins fréquentes.
Aucune différence clinique netten’a été identifiée entre les phénotypes LOFA et vLOFA, si ce n’est la tendance à une meilleure préservation fonctionnelle dans les formes les plus tardives. Ces patients conservent en moyenne plus longtemps la marche autonome et présentent un handicap moteur moins sévère.
Sur le plan moléculaire
Les formes tardives sont associées à une expansion GAA plus courte sur l’allèle le plus petit, ce qui expliquerait la corrélation inverse entre la taille de l’expansion et la sévérité du phénotype.
Diagnostic différentiel
Plusieurs affections neurologiques peuvent mimer le tableau clinique de FA, rendant le diagnostic différentiel essentiel, en particulier dans les tardives.
Les principales pathologies à évoquer sont :
• les neuropathies héréditaires sensitivo-motrices (maladie de Charcot-Marie-Tooth),
• l’ataxie-télangiectasie,
• les ataxies avec apraxie oculomotrice,
• l’ataxie par déficit en vitamine E
• et l’abêtalipoprotéinémie.
Ces affections partagent avec l’ataxie de Friedreich une ataxie progressive et une atteinte proprioceptive, mais elles s’en distinguent par certains éléments cliniques, biologiques ou génétiques présentés dans le tableau 1.
La distinction repose sur l’association de critères cliniques spécifiques (mouvements oculaires anormaux, télangiectasies, spasticité), d’examens biologiques ciblés (vitamine E, lipides, alpha-fœtoprotéine, électrophorèse des lipoprotéines), de l’électroneuromyogramme, et surtout de la confirmation génétique.
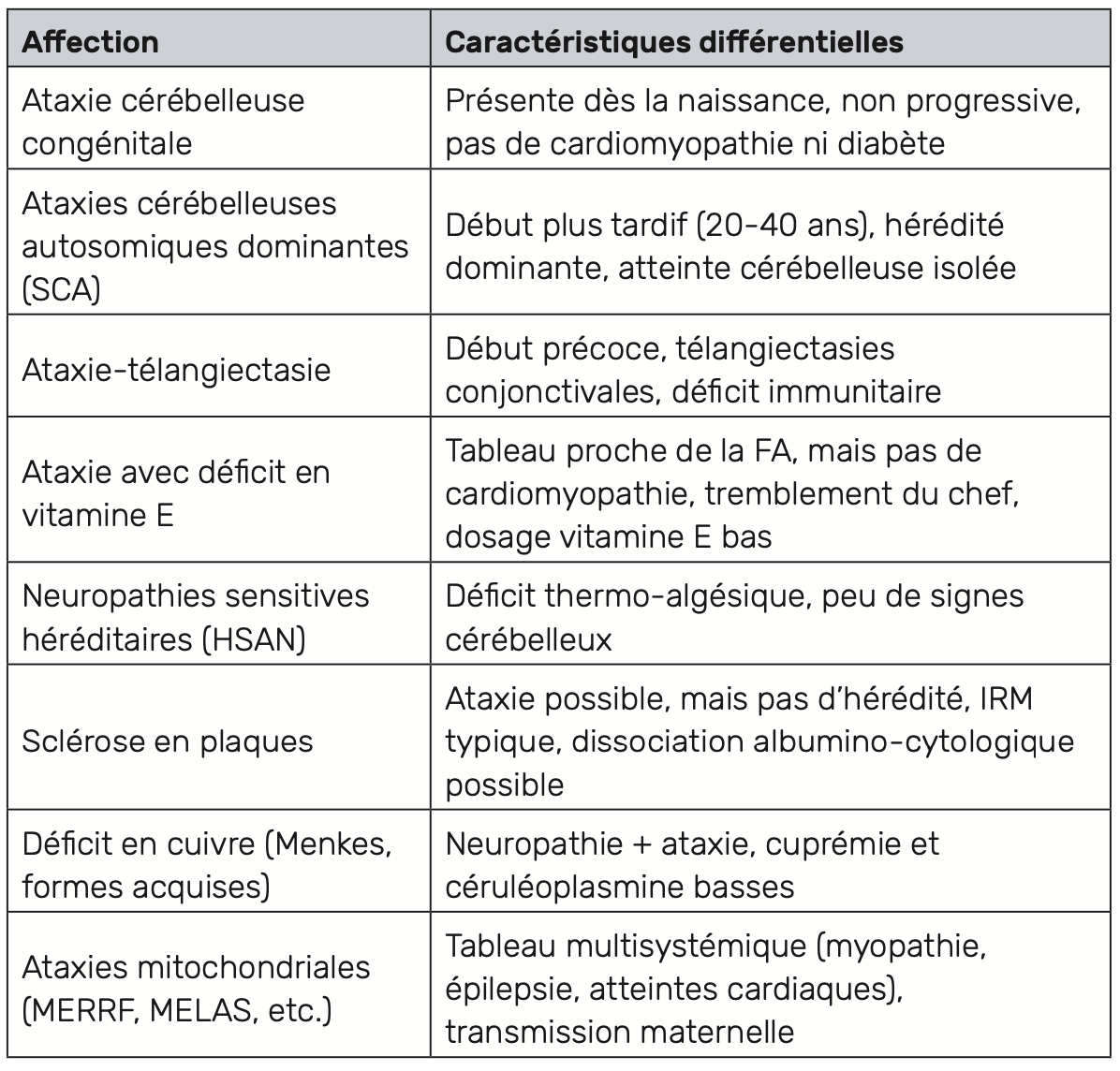
Tableau 1 – Diagnostic différentiel.
Examens complémentaires
Les examens complémentaires dans la FA ont un double objectif : confirmer le diagnostic et assurer le suivi évolutif et systémique de la maladie.
Examens diagnostiques
• Aux stades précoces, les IRM cérébrale et médullaire sont souvent normales. À un stade plus avancé, elles mettent en évidence une atrophie médullaire, prédominant au niveau cervical, ainsi qu’une atteinte des pédoncules cérébelleux supérieurs, contrastant avec un volume cérébelleux globalement préservé. Des études récentes ont montré que la surface de la moelle cervicale est corrélée à la sévérité clinique évaluée par les échelles spécifiques de la FRDA (FARS, SARA).
• L’électroneuromyogramme (ENMG) révèle une polyneuropathie axonale sensitive, souvent symétrique, avec abolition ou diminution des potentiels sensitifs distaux. L’atteinte motrice est inconstante et généralement modérée.
• Le diagnostic de certitude repose sur la génétique moléculaire, avec la mise en évidence d’une expansion de la triplet GAA dans l’intron 1 du gène FXN. Dans 2 à 5 % des cas, une mutation ponctuelle est retrouvée sur l’autre allèle, justifiant un séquençage complet du gène en cas de résultat partiellement ambigu.
Examens de suivi systémique
Le suivi est multidisciplinaire et inclut :
• une surveillance métabolique : dépistage régulier du diabète (glycémie à jeun, HbA1c) et du profil lipidique, compte tenu du risque d’intolérance au glucose et de surpoids ;
• une évaluation cardiologique : recherche systématique d’une cardiomyopathie hypertrophique ou d’une dysfonction ventriculaire, par ECG et échocardiographie, complétés au besoin par une IRM cardiaque permettant d’évaluer la fibrose myocardique (séquences LGE, T1 mapping) ;
• suivi orthopédique et rééducatif : évaluation de la scoliose, des déformations du pied et de la spasticité.
Examens émergents et biomarqueurs
De nouveaux outils d’évaluation sont à l’étude pour affiner le suivi longitudinal et la mesure de la progression :
• l’IRM quantitative : mesures de diffusion, de relaxométrie T1/T2* et de susceptibilité magnétique, permettant de quantifier la dégénérescence médullaire et cérébelleuse ;
• la spectroscopie par résonance magnétique (SRM) : exploration du métabolisme neuronal (réduction du N-acétyl-aspartate dans les pédoncules cérébelleux et la moelle) ;
• les potentiels évoqués somesthésiques et moteurs : évaluation de la conduction au sein des voies longues sensitives et motrices, utiles dans le suivi précoce ;
• les biomarqueurs circulants : la frataxine sanguine ou leucocytaire est un marqueur prometteur corrélé à la sévérité phénotypique et à la taille de l’expansion GAA ; des biomarqueurs mitochondriaux et inflammatoires sont également en cours de validation.
Évolution
La FA est une maladie neurodégénérative lentement progressive, mais dont le pronostic reste globalement sombre à long terme.
Évolution des symptômes
La progression neurologique est continue, avec une aggravation moyenne du score SARA d’environ +0,86 point par an, plus rapide dans les formes à début précoce (+1,04 point/an) [11].
L’évolution des symptômes de la maladie de FA est présentée dans la figure 5. Elle montre que tous les symptômes s’aggravent de façon progressive avec le temps. La faiblesse musculaire, initialement distale, s’accentue progressivement et contribue à l’aggravation de l’instabilité posturale et à la perte de la marche autonome. Les troubles de l’équilibre s’aggravent.
La majorité des patients deviennent dépendants du fauteuil roulant à l’âge adulte jeune [12].
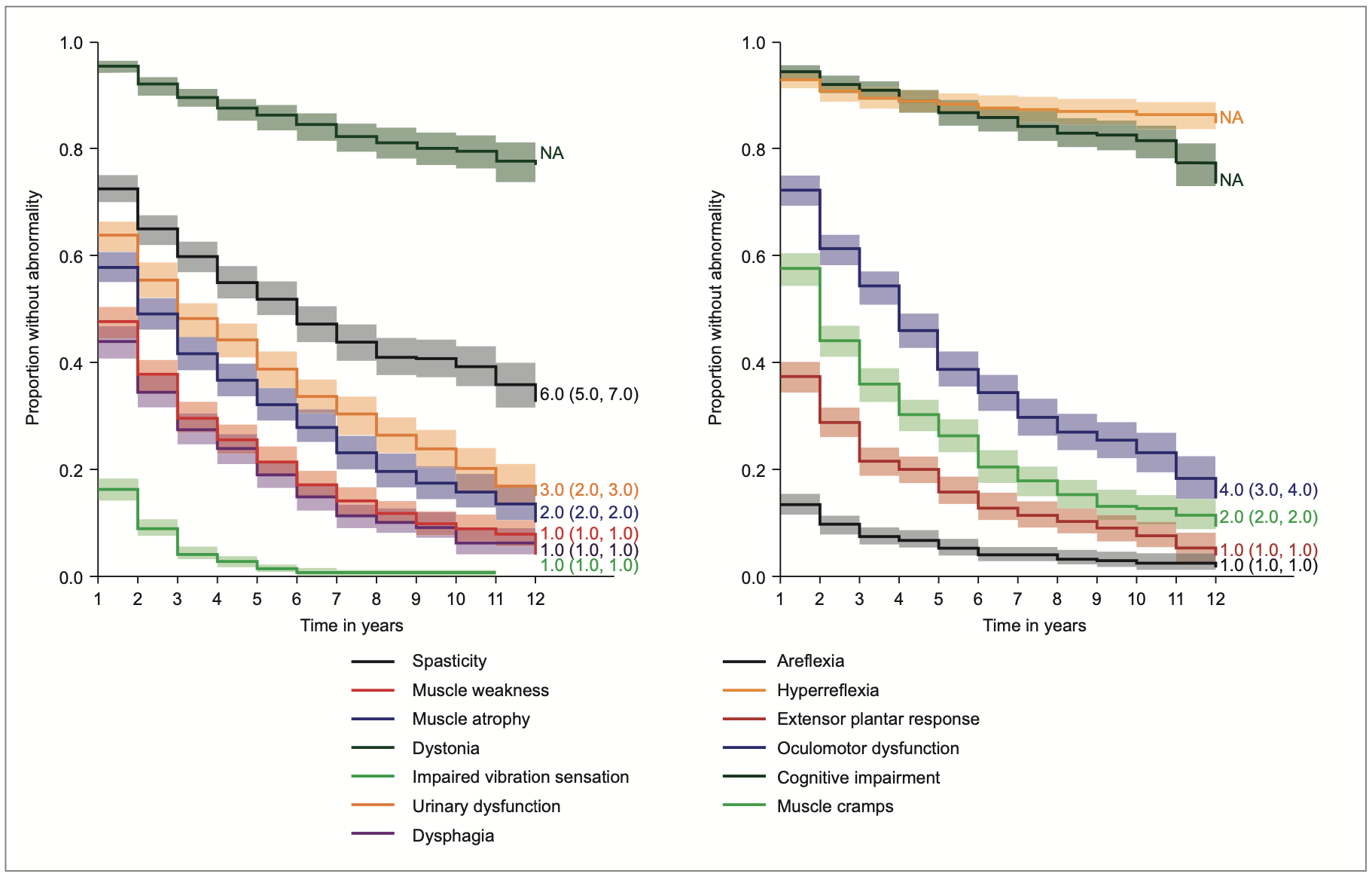
Figure 5 – Proportion de patients indemnes de symptômes de la maladie de FA avec le temps [13].
Mortalité
L’espérance de vie moyenne est estimée entre 35 et 40 ans (36,5 ans selon [7]), mais une variabilité importante est observée, certains patients survivant jusqu’à la septième, voire la huitième décennie.
Les principales causes de décès sont cardiaques, dans environ 60 % des cas, dominées par l’insuffisance cardiaque congestive et les troubles du rythme. Les causes non cardiaques, notamment la pneumopathie d’inhalation liée à la dysphagie (28 % des cas), représentent la deuxième cause de mortalité [4]. La dysphagie est en effet très fréquente, rapportée chez 69 à 98 % des patients, favorisant les complications respiratoires à un stade avancé.
Facteurs pronostiques défavorables
Certains facteurs pronostiques défavorables ont été clairement identifiés :
• âge de début précoce,
• taille importante de l’expansion GAA (notamment sur l’allèle GAA1),
• présence d’une cardiomyopathie ou d’un diabète.
Sur le plan cardiaque, la diminution de la fraction d’éjection ventriculaire gauche et l’augmentation de l’indice de masse ventriculaire gauche constituent des marqueurs prédictifs d’évolution vers l’insuffisance cardiaque et d’une mortalité accrue.
Au-delà des complications neurologiques et cardiaques, l’évolution de la FA s’accompagne souvent d’atteintes endocriniennes, respiratoires et musculosquelettiques, affectant la qualité de vie. Elle exerce également une charge psychologique, cognitive, sociale et familiale importante, soulignant la nécessité d’une prise en charge pluridisciplinaire continue.
Prise en charge et suivi
La prise en charge de FA doit être multidisciplinaire et idéalement coordonnée au sein d’un centre expert maladies rares.
Elle repose sur un suivi régulier, adapté à la progression de la maladie et à ses complications multi-systémiques :
• neurologique : suivi semestriel à annuel, évaluation motrice standardisée (SARA ou mFARS), kinésithérapie de maintien, orthophonie, et prise en charge ciblée des troubles vésico-sphinctériens ou du sommeil ;
• cardiaque : électrocardiogramme et échocardiographie tous les 1 à 2 ans, dépistage précoce des cardiomyopathies et troubles du rythme ;
• métabolique : dosage annuel de l’HbA1c et de la glycémie pour dépister un diabète ou une intolérance au glucose ;
• nutritionnel : surveillance du poids, dépistage des fausses routes et adaptation des apports nutritionnels.
• ophtalmologique et ORL : bilans visuels et auditifs tous les 1 à 2 ans.
L’approche doit être globale et personnalisée, combinant soins médicaux, rééducation fonctionnelle, accompagnement psychologique, et soutien social et éducatif, dans l’objectif d’améliorer la qualité de vie et de préserver l’autonomie. L’arrivée de l’omavéloxolone, premier traitement spécifique ayant obtenu une AMM, constitue une avancée thérapeutique majeure, mais ne se substitue pas à la nécessité d’un suivi intégré et coordonné.
Défis
Les principaux défis actuels résident dans :
• la coordination du parcours de soins,
• la transition pédiatrique-adulte,
• la prévention de l’épuisement des patients et des familles,
• et le renforcement du maillage territorial des centres experts maladies rares.
Traitement
À ce jour, aucun traitement curatif de la FA n’est disponible. La prise en charge repose sur des approches symptomatiques, rééducatives et cardio-métaboliques, associées à un suivi multidisciplinaire.
Traitements conventionnels
• Antioxydants mitochondriaux : l’idébénone et la coenzyme Q10 ont été largement étudiés entre 2000 et 2010 (essai MICONOS, 232 patients), sans démonstration d’un bénéfice neurologique significatif. Les effets cardiaques restent controversés.
• Prise en charge cardiaque : selon les recommandations usuelles, inhibiteurs de l’enzyme de conversion (IEC), antagonistes des récepteurs de l’angiotensine II (ARA2), bêtabloquants, diurétiques en cas d’insuffisance cardiaque, défibrillateur si FEVG < 35 %, et anticoagulants en cas de fibrillation auriculaire.
• Approches non médicamenteuses : kinésithérapie, orthophonie, appareillage et ventilation assistée en cas d’apnées du sommeil.
Traitement spécifique : omavéloxolone
L’omavéloxolone est le premier traitement spécifique de FA approuvé par la FDA en 2023 et la Commission européenne en 2024 pour les patients âgés de plus de 16 ans.
Il s’agit d’un activateur de la voie Nrf2 (se référer à la figure 2 pour identifier Nrf2), régulant l’expression de gènes antioxydants (NQO1, SRXN1, TXNRD1) et améliorant la fonction mitochondriale.
Résultats
L’essai multicentrique MOXIe a montré, après 48 semaines de traitement (160 mg/jour), une amélioration moyenne de +1,55 point sur l’échelle mFARS, contre une dégradation de -0,85 point dans le groupe placebo. L’étude d’extension ouverte sur 3 ans a confirmé un maintien du bénéfice clinique, comparativement à l’histoire naturelle observée dans le registre FACOMS. Cet effet semble se maintenir à 5 ans [14].
Les effets indésirables les plus fréquents sont une élévation transitoire des transaminases (souvent réversible en moins de 12 semaines) et une augmentation du cholestérol, sans atteinte hépatique significative.
L’amélioration fonctionnelle observée est comparable au déclin habituellement constaté sur 2 ans d’évolution naturelle, ce qui en fait une avancée majeure dans la prise en charge.
Cependant, les limites des essais incluent le faible effectif, l’absence de données pédiatriques et la nécessité d’un suivi prolongé.
Perspectives thérapeutiques
Les recherches en cours explorent :
• la modulation transcriptionnelle du gène FXN,
• les thérapies géniques et ARN antisens,
• les approches cellulaires et pharmaco-chaperons visant à restaurer la production de frataxine.
|
À retenir |
|
• • • • |
L’auteur déclare eavoir réalsié des présentations sur l’ataxie de Friedreich à l occasion de congrès à la demande de Biogen.
Bibliographie
1. Buesch K, Zhang R. A systematic review of disease prevalence, health-related quality of life, and economic outcomes associated with Friedreich’s ataxia. Curr Med Res Opin 2022 ; 38 : 1739-49.
2. Campuzano V, Montermini L, Moltò MD et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996 ; 271 : 1423-27.
3. Delatycki MB, Williamson R, Forrest SM. Friedreich ataxia: an overview. J Med Genet 2000 ; 37 : 1-8.
4. Reetz K, Lischewski SA, Dogan I et al. Friedreich’s ataxia—a rare multisystem disease. Lancet Neurol 2025 ; 24 : 614-24.
5. González-Cabo P, Palau F. Mitochondrial pathophysiology in Friedreich’s ataxia. J Neurochem 2013 ; 126 : 53-64.
6. Petrillo S, Piermarini E, Pastore A et al. Nrf2 as a potential therapeutic target in Friedreich’s ataxia. Trends Pharmacol Sciences 2019 ; 40 : 256-71.
7. Reetz K, Dogan I, Hilgers RD et al. Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 2 year cohort study. Lancet Neurol 2016 ; 15 : 1346-54.
8. Reetz K, Dogan I, Hohenfeld C et al. Nonataxia symptoms in Friedreich Ataxia: report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 2018 ; 91 : e91730.
9. Corben LA, Collins V, Milne S et al. Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet J Rare Dis 2022 ; 17 : 415.
10. Cook A, Giunti P. Friedreich’s ataxia: clinical features, pathogenesis and management. Br Med Bull 2017 ; 124 : 19-30.
11. Reetz K, Dogan I, Hilgers RD et al. Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol 2021 ; 20 : 362-72.
12. Rummey C, Farmer JM, Lynch DR. Predictors of loss of ambulation in Friedreich’s ataxia. EClinicalMedicine 2020 ; 18: 100213.
13. Lischewski SA, Dogan I, Giunti P et al. Analysis of a modified version of the inventory of non-ataxia signs over 12 years in patients with Friedreich’s ataxia in the EFACTS study. Mov Disord. 2025 ; Online ahead of print.
14. Nachbauer W, Lynch D, Delatycki M et al. 234P Long-term use of omaveloxolone in patients with Friedreich ataxia: up to 5 years of natural history propensity score matching from the MOXIe OLE. Neuromuscul Disord 2025 ; 53 : 106043.