
Résumé
• L’association neuropathie et mouvements anormaux ou ataxie cérébelleuse concerne de très nombreuses pathologies.
• Les points-clés du raisonnement diagnostique, dans ce cadre, sont la caractérisation du profil clinico-électrique de la neuropathie, la caractérisation des mouvements anormaux, la recherche de signes associés neurologiques et extra-neurologiques.
• Le raisonnement diagnostique doit s’attacher à rechercher en priorité les causes traitables.
• De nombreuses pathologies neurométaboliques sont responsables de l’association neuropathie et mouvements anormaux et peuvent être dépistées par des examens biologiques simples (biomarqueurs).
Abstract
Movement disorders and peripheral neuropathies
• Numerous diseases can present with the association of a peripheral neuropathy with movement disorders or cerebellar ataxia.
• The most important steps of clinical reasoning in this setting are the characterization of the clinical and electrical profile of the neuropathy, the semiology of movement disorders, and the search for additional neurological and extra-neurological signs.
• Diagnosis of treatable causes should always be prioritized.
• Metabolic disorders are an important cause of movements disorders and peripheral neuropathy, and the clinician should be aware of simple biological biomarkers for these diseases.
L’objectif de cette revue est de présenter un aperçu non exhaustif de la démarche diagnostique et des grands cadres étiologiques à évoquer chez des patients présentant l’association de mouvements anormaux (ou d’une ataxie cérébelleuse) et d’une neuropathie périphérique. De très nombreuses pathologies, notamment génétiques et neurométaboliques, peuvent être responsables de cette association [1].
Les éléments fondamentaux du raisonnement diagnostique dans cette situation sont :
• la caractérisation du profil clinico-électrique de la neuropathie (neuronopathie sensitive, neuropathie démyélinisante, neuropathie motrice pure ou à prédominance motrice, neuropathie axonale sensitivo-motrice). Cette étape nécessite un examen clinique rigoureux ainsi que la réalisation d’un ENMG des quatre membres, comprenant une étude de la conduction motrice étagée, une étude de la conduction sensitive, un EMG de détection. Plus le profil de la neuropathie est spécifique, plus sa valeur d’orientation diagnostique sera importante ;
• la caractérisation clinique des mouvements anormaux (ataxie cérébelleuse, tremblement, syndrome parkinsonien, dystonie, chorée, myoclonies, etc.) ;
• l’âge de début et le mode d’apparition de la symptomatologie ;
• la présence d’antécédents familiaux, la notion d’une censure et/ou d’une consanguinité familiale ;
• les signes cliniques associés, neurologiques et extra-neurologiques (ophtalmologiques, cutanés, etc.)
• et les éléments paracliniques (IRM cérébrale notamment).
La caractérisation de ces différents points permettra d’étoffer les termes de l’équation diagnostique et de limiter le nombre de diagnostics différentiels par un raisonnement “en entonnoir”. Par exemple, l’association neuropathie périphérique et ataxie cérébelleuse est fréquente et non spécifique, mais l’association d’une neuronopathie sensitive à une ataxie cérébelleuse, un tremblement du chef, une rétinite pigmentaire, débutant dans l’enfance, dans un contexte de consanguinité familiale, va faire évoquer très rapidement une ataxie par déficit en vitamine E (AVED). La démarche diagnostique doit également être guidée par la recherche prioritaire des pathologies accessibles à un traitement, d’autant que beaucoup d’entre elles peuvent être dépistées par des examens simples [2].
Cette revue ne pouvant être exhaustive, elle mettra l’accent sur les pathologies les plus fréquentes, les pathologies traitables, et les pathologies pouvant être diagnostiquées à l’âge adulte. Ne seront pas citées les pathologies où la neuropathie est rare et n’aide en général pas le diagnostic (maladie de Wilson par exemple). La caractérisation du profil clinico-électrique de la neuropathie étant un élément clé dans le diagnostic, nous utiliserons les grands profils de neuropathies périphériques comme plan de la revue.
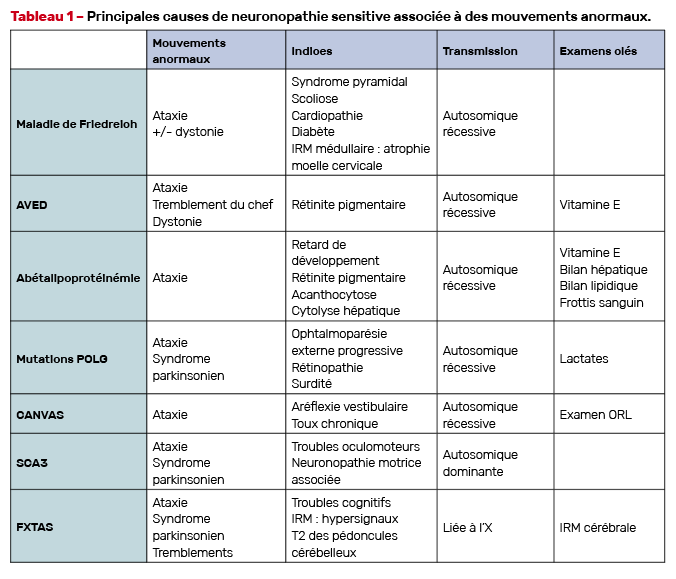
Neuronopathies sensitives (Tab. 1)
Les neuronopathies sensitives se caractérisent par une neuropathie sensitive diffuse, non longueur dépendante. L’association neuronopathie sensitive et mouvements anormaux ou ataxie cérébelleuse est une situation clinique relativement fréquente et confortable pour le clinicien, avec quelques étiologies clés à bien connaître.
La maladie de Friedreich
La maladie de Friedreich, liée à des mutations homozygotes du gène de la frataxine, se révèle souvent par des troubles de la marche en rapport avec une neuropathie sensitive, associée à un syndrome cérébelleux, un syndrome pyramidal, parfois une dystonie. Des formes de début tardif sont décrites.
L’ataxie avec déficit en vitamine E (AVED)
L’ataxie avec déficit en vitamine E débute en général dans l’enfance ou l’adolescence. Elle se manifeste par une ataxie, une aréflexie, des troubles proprioceptifs, des mouvements anormaux (ataxie cérébelleuse, tremblement touchant souvent le chef, dystonie) [3]. Elle peut s’accompagner d’une rétinite pigmentaire. Il s’agit d’une pathologie traitable, justifiant un traitement substitutif à vie par vitamine E.
L’abétalipoprotéinémie
L’abétalipoprotéinémie ou maladie de Bassen-Kornzweig se manifeste en général par une neuronopathie sensitive associée à une ataxie cérébelleuse, une rétinite pigmentaire, un syndrome de malabsorption, des troubles cognitifs. Sur le plan biologique, une acanthocytose ou une cytolyse hépatique peuvent être notées. Il s’agit comme l’AVED d’une pathologie traitable (régime dépourvu de graisses à chaîne longue et supplémentation en vitamines liposolubles dont la vitamine E).
Pré-mutation X fragile (FXTAS)
Le syndrome tremblement-ataxie lié à une prémutation du gène FMR1 (FXTAS) débute à l’âge adulte, après 50 ans, et est caractérisé par l’association variable d’un tremblement d’action, d’une ataxie cérébelleuse, d’un syndrome parkinsonien, d’un syndrome dysexécutif. Sur le plan périphérique, il peut se manifester par une neuronopathie sensitive ou une polyneuropathie axonale à prédominance sensitive, souvent en arrière-plan. L’IRM cérébrale peut montrer des hypersignaux T2 très évocateurs notamment au niveau des pédoncules cérébelleux. Ce diagnostic est important car il a des implications majeures en termes de conseil génétique (risque de syndrome de l’X fragile dans la famille) [4].
Les pathologies mitochondriales
Les pathologies mitochondriales et notamment les mutations du gène POLG peuvent se traduire par une neuronopathie sensitive associée à une ataxie cérébelleuse, un syndrome parkinsonien, une dystonie. Les indices cliniques sont la présence d’une ophtalmoparésie externe progressive, d’une rétinopathie, de troubles auditifs [5]. Le taux de lactates peut être élevé.
SCA3
Les formes tardives de SCA3 ou maladie de Machado-Joseph se manifestent par une ataxie cérébelleuse, un syndrome parkinsonien parfois au premier plan [6], une neuronopathie sensitive parfois associée à une neuronopathie motrice (double neuronopathie).
Le syndrome CANVAS
Le syndrome CANVAS associe syndrome cérébelleux, neuronopathie sensitive et aréflexie vestibulaire. Il s’agit d’une cause fréquente de neuronopathie sensitive survenant à l’âge adulte. Son substrat génétique a récemment été identifié (mutations bialléliques du gène RFC1).
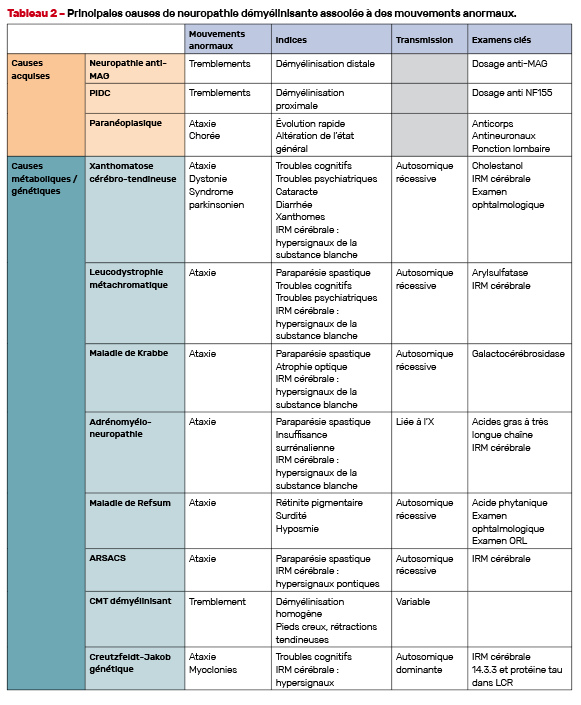
Neuropathies démyélinisantes (Tab. 2)
Le contexte (âge de début, antécédents familiaux), le mode d’apparition de la symptomatologie, la présence de signes associés, la présence ou non d’une dissociation électro-clinique et la caractérisation plus précise du profil de neuropathie démyélinisante (homogène, inhomogène avec blocs de conduction, à prédominance distale, axono-démyelinisante, etc.) permettront de s’orienter facilement entre les causes acquises (en premier lieu polyradiculonévrite chronique et neuropathie anti-MAG qui peuvent être associées à un tremblement), les formes démyélinisantes de maladie de Charcot-Marie-Tooth avec tremblements, et les neuropathies associées aux pathologies métaboliques.
Causes acquises
Les neuropathies anti-MAG s’accompagnent souvent d’un tremblement d’attitude et d’action distal des membres supérieurs [7]. Les polyneuropathies inflammatoires démyélinisantes chroniques (PIDC) peuvent également s’accompagner d’un tremblement, particulièrement fréquent dans les PIDC associés aux anticorps anti-neurofascine 155, avec parfois des signes cérébelleux surajoutés [8]. Dans ces différents cas, on parle de tremblement neuropathique car on suppose un lien physiopathologique entre la neuropathie et la survenue du tremblement.
Dans le cas d’une neuropathie axono-démyélinisante d’évolution rapide, associée à un syndrome cérébelleux, éventuellement d’autres mouvements anormaux (chorée, dystonie), on évoquera une neuropathie paranéoplasique.
Causes métaboliques ou génétiques
Parmi les pathologies neurométaboliques, la xanthomatose cérébro-tendineuse est à évoquer en priorité. Cette maladie débute dans l’enfance, mais il n’est pas rare que le diagnostic soit posé à l’âge adulte. Elle peut se manifester par une ataxie, une dystonie, un syndrome parkinsonien, souvent associés à une paraparésie spastique, des troubles cognitifs et psychiatriques. Sur le plan extra-neurologique, une cataracte débutant dans l’enfance, une diarrhée chronique, la présence de xanthomes cutanés, une hépatopathie sont des indices diagnostiques importants. L’IRM cérébrale met en évidence des hypersignaux T2 encéphaliques touchant notamment les faisceaux cortico-spinaux, la substance blanche périventriculaire, les noyaux dentelés. Le diagnostic est suspecté par une diminution du cholestanol plasmatique, et confirmé par l’analyse du gène CYP27A1. Ce diagnostic est incontournable car cette pathologie fait l’objet d’un traitement qui peut ralentir, voire stopper, l’évolution de la maladie (acide chénodésoxycholique) [9].
La leucodystrophie métachromatique et la maladie de Krabbe peuvent également se manifester par une ataxie cérébelleuse, le plus souvent associée à une paraparésie spastique. Ces pathologies débutent dans l’enfance mais certaines formes tardives sont diagnostiquées à l’âge adulte.
Les maladies péroxysomales peuvent également se manifester par une neuropathie démyélinisante associée à une ataxie et peuvent être dépistées par des examens biologiques simples (acides gras à très longues chaînes, acide phytanique, acide pristanique).
C’est le cas notamment de l’adrénomyéloneuropathie, qui se manifeste souvent sur le plan périphérique par une neuropathie axono-démyélinisante. La présence d’une atteinte neurosensorielle (surdité de perception, rétinite pigmentaire) fera évoquer une maladie de Refsum, accessible à un traitement (régime pauvre en acide phytanique).
L’ataxie spastique autosomique récessive de Charlevoix-Saguenay (ARSACS) se manifeste par une ataxie cérébelleuse et une paraparésie spastique, associées à une neuropathie le plus souvent axono-démyelinisante.
Des formes démyélinisantes de maladie de Charcot-Marie-Tooth (notamment le CMT1) peuvent s’associer à un tremblement neuropathique.
Des neuropathies démyélinisantes, parfois avec blocs de conduction, ont été décrites dans certains cas de maladies de Creutzfeldt-Jakob, notamment dans des formes génétiques (mutations E200K du gène PRNP).
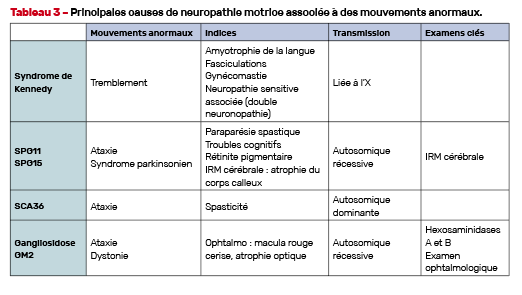
Neuropathies motrices pures ou à prédominance motrice (Tab. 3)
Le syndrome de Kennedy
Le syndrome de Kennedy ou amyotrophie spinobulbaire s’accompagne souvent d’un tremblement. Un tremblement d’attitude a également été rapporté dans différentes formes d’amyotrophie spinale et dans le syndrome d’Hirayama.
SPG11 et SPG15
Les paraparésies spastiques autosomiques récessives SPG11 et SPG15 peuvent s’accompagner d’une neuropathie motrice. Le phénotype clinique dans ces pathologies est souvent complexe, associant paraparésie spastique, ataxie cérébelleuse, troubles cognitifs, syndrome parkinsonien. L’IRM cérébrale retrouve fréquemment une atrophie du corps calleux ainsi que des hypersignaux T2 au niveau des cornes frontales des ventricules latéraux (signe des “oreilles de lynx”).
SCA36
Le SCA36 se manifeste également souvent par une neuropathie à prédominance motrice, avec une atrophie de la langue, des fasciculations, une neuropathie motrice distale, associée à une ataxie cérébelleuse et une spasticité des membres inférieurs.
Gangliosidose
La gangliosidose GM2, liée à des mutations du gène de l’hexoaminidase, peut se manifester dans ses formes tardives par une neuropathie à prédominance motrice proche ressemblant à une amyotrophie spinale, associée à une ataxie cérébelleuse, une dystonie, une chorée-athétose, des myoclonies.
Neuropathies axonales sensitivo-motrices (Tab. 4)
Il s’agit de la situation clinique la plus difficile du fait du nombre extrêmement important d’étiologies possibles, seules certaines causes seront donc abordées. Ce profil de neuropathie étant moins spécifique, le type de mouvements anormaux et les signes associés vont être particulièrement pertinents pour guider la démarche diagnostique. Des pathologies citées dans les précédentes sections peuvent se manifester également par une neuropathie axonale sensitivo-motrice, c’est le cas notamment de la maladie de Friedreich, des mutations POLG, du FXTAS.
Causes acquises
Parmi les causes acquises, les neuropathies paranéoplasiques sont à évoquer dans ce contexte. Notamment, les neuropathies liées aux anti-CRMP5/CV2 se manifestent souvent par une neuropathie axonale douloureuse, une ataxie cérébelleuse, des mouvements choréiques [10]. La maladie cœliaque peut être responsable à la fois d’une neuropathie, souvent à prédominance sensitive, parfois douloureuse, et d’une ataxie cérébelleuse. Elle peut également se manifester par des myoclonies, y compris du voile du palais.
Causes métaboliques ou génétiques
De nombreuses ataxies spinocérébelleuses autosomiques dominantes peuvent être associées à une neuropathie axonale sensitivo-motrice. Nous pouvons par exemple citer le SCA1, le SCA2, le SCA4, le SCA7, le SCA8, le SCA10, le SCA12, le SCA18, le SCA23, le SCA25.
Parmi les ataxies cérébelleuses récessives, le groupe des ataxies avec apraxie oculomotrice, qui comprend l’ataxie télangiectasie, l’ataxie avec apraxie oculomotrice de type 1 (AOA1), et de type 2 (AOA2) se manifeste cliniquement par une ataxie cérébelleuse souvent sévère, débutant dans l’enfance, pouvant être associée à d’autres mouvements anormaux (dystonie, chorée, myoclonies, syndrome parkinsonien). Ces pathologies sont souvent associées à une neuropathie axonale sensitivo-motrice parfois sévère. L’association à une apraxie oculomotrice est caractéristique. Il existe une variabilité phénotypique très importante, notamment dans l’ataxie-télangiectasie. L’élévation de l’alpha-fœto-protéine est un biomarqueur important de ce groupe de pathologies. Le diagnostic d’ataxie-télangiectasies est particulièrement important car une surveillance est recommandée du fait d’un risque élevé de cancers (pathologie de réparation de l’ADN).
De nombreuses pathologies mitochondriales, comme les mutations POLG précédemment citées, peuvent s’accompagner d’une neuropathie sensitivo-motrice, avec ataxie cérébelleuse et syndrome parkinsonien. Le syndrome NARP lié aux mutations du gène MT-ATP6 de l’ADN mitochondrial se manifeste par une neuropathie axonale sensitivo-motrice, une ataxie, une rétinite pigmentaire, une cardiomyopathie. L’ataxie cérébelleuse est également fréquente dans le MERRF (myoclonic epilepsy with ragged red fibers) et le SPG7. Une dystonie associée à une neuropathie axonale fait essentiellement évoquer une pathologie mitochondriale (mutation POLG, Twinkle, syndrome de Leigh, neuropathie optique de Leber). L’association d’une neuropathie à des myoclonies corticales fait également évoquer une pathologie mitochondriale, notamment un syndrome MELAS (mitochondrial myopathy with encephalopathy, lactic acidosis and stroke-like episodes), MERRF ou NARP. Les mutations POLG sont une des rares causes d’ataxie progressive avec tremblement du voile du palais.
Une chorée généralisée associée à une neuropathie périphérique fera évoquer le cadre des neuro-acanthocytoses : la chorée-acanthocytose autosomique récessive liée au gène VPS13A et le syndrome de McLeod lié à l’X (gène XK). La présence d’acanthocytes sur le frottis sanguin et une augmentation des CPK sont des indices importants pour le diagnostic.
Chez un patient présentant un syndrome parkinsonien au premier plan, de début précoce, associé à une neuropathie périphérique, on peut évoquer une maladie de Kufor-Rakeb ou une mutation du gène PLA2G6. n
Correspondance
cecile.delorme@aphp.fr
L’auteure déclare ne pas avoir de lien d’intérêt avec le sujet de l’article.
Bibliographie
1. Rossor AM, Carr AS, Devine H et al. Peripheral neuropathy in complex inherited diseases: an approach to diagnosis. J Neurol Neurosurg Psychiatry 2017 ; 88 : 846‑63.
2. Anheim M, Tranchant C. Peripheral neuropathies associated with hereditary cerebellar ataxias. Rev Neurol 2011 ; 167 : 72‑6.
3. Pradeep S, Ali T, Guduru Z. Ataxia with Vitamin E Deficiency with Predominant Cervical Dystonia. Mov Disord Clin Pract 2020 ; 7 : 100‑3.
4. Hall DA, Berry-Kravis E. Fragile X syndrome and fragile X-associated tremor ataxia syndrome. Handb Clin Neurol 2018 ; 147 : 377‑91.
5. Tchikviladzé M, Gilleron M, Maisonobe T et al. A diagnostic flow chart for POLG-related diseases based on signs sensitivity and specificity. J Neurol Neurosurg Psychiatry 2015 ; 86 : 646‑54.
6. Escorcio Bezerra ML, Pedroso JL, Spinola Pinheiro D et al. Pattern of peripheral nerve involvement in Machado-Joseph disease: neuronopathy or distal axonopathy? A clinical and neurophysiological evaluation. Eur Neurol 2013 ; 69 : 129‑33.
7. Ahlskog MC, Kumar N, Mauerman ML et al. IgM-monoclonal gammopathy neuropathy and tremor: A first epidemiologic case control study. Parkinsonism & Related Disorders 2012 ; 18 : 748‑52.
8. Bailly L, Mongin M, Delorme C et al. Tremor Associated with Chronic Inflammatory Demyelinating Polyneuropathy and Anti-Neurofascin-155 Antibodies. Tremor Other Hyperkinet Mov 2018 ; 8 : 606.
9. Degos B, Nadjar Y, Amadir M et al. Natural history of cerebrotendinous xanthomatosis: a paediatric disease diagnosed in adulthood. Orphanet J Rare Dis 2016 ; 11 : 41.
10. Dubey D, Lennon VA, Gadoth A et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology 2018 ; 90 : e103-10.