
7 et 8 février 2020 à Paris
La Société francophone du nerf périphérique a tenu ses 24es Journées annuelles les 7 et 8 février derniers, dans le très joli cadre de la Maison de la Chimie à Paris. Comme tous les ans, elles ont rassemblé tous ceux primitivement intéressés par ces pathologies nerveuses périphériques, aussi bien cliniciens que chercheurs. Elles ont été particulières par la tenue, pour la première fois, d’une réunion commune avec nos collègues et amis de la Société britannique de neuropathies périphériques (BPNS), et ce, malgré la concomitance du Brexit.
Grâce à la complémentarité de nos deux sociétés, cette journée partagée a permis d’importantes mises au point cliniques sur différents thèmes d’actualité y compris thérapeutique, aussi bien dans le champ génétique que dysimmunitaire. Une session spécifique a été dédiée à une approche moderne de la douleur, très appréciée par les participants. La journée s’est conclue par un authentique ‘crush’, un match opposant deux équipes de jeunes neurologues français et anglais, autour de quizz cliniques, match que les jeunes collègues français ont eu l’élégance de perdre au profit des invités d’outre-Manche. Mais ce ne sera que partie remise.
La journée du 8 février avait, comme tous les ans, une coloration de formation médicale continue, alternant mises au point didactiques, revues de la bibliographie 2019 et observations interactives – pas toujours faciles.
Vous trouverez dans la lecture des petites synthèses à suivre un condensé forcément arbitraire du contenu de ces journées, dont le succès ne se dément pas au fil des ans. Rendez-vous l’année prochaine !
Yann Péréon
Sélection de communications : CANVAS, imagerie du nerf et effet placebo
par Armelle Magot
Quelle est la surprise génétique de cette année écoulée ?
La surprise est venue de la découverte du gène responsable d’une neuropathie génétique rare, le syndrome CANVAS (Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome).
Ce syndrome, connu depuis de nombreuses années, mais échappant aux biologistes moléculaires jusqu’à présent, associe une neu ropathie sensitive avec une toux chronique non expliquée, souvent explorée antérieurement. Il peut s’y ajouter une atteinte vestibulaire ou cérébelleuse.
Le Dr Cortese (Londres) a présenté les résultats de son équipe qui, à partir de 29 patients issus de 11 familles, a mis en évidence une expansion récessive AAGGG dans l’intron 2 du gène RFC1.
- Cinquante-six patients ont finalement été phénotypés dans leur cohorte : l’âge moyen de début est de 54 ans.
- La plainte la plus fréquente est le trouble de l’équilibre qui est reporté chez 84 % des patients, classiquement aggravé dans le noir.
- Chez 37 % des patients, la plainte d’une toux chronique est notée, qui peut parfois précéder de plusieurs années la neuropathie, sans étiologie ORL ou pneumologique retrouvée.
- Les patients ont des signes d’une neuropathie à grosses fibres dans tous les cas, 80 % ont des signes cérébelleux et 54 % ont une aréflexie vestibulaire bilatérale.
- Chez 23 % des patients, on retrouve une atteinte dysautonomique, avec notamment des troubles urinaires et de l’élimination fécale.
- L’électroneuromyogramme montre systématiquement une neuropathie sensitive des grosses fibres et 83 % des patients ont une atrophie cérébelleuse à l’IRM ou au scanner.
Pour résumer, regardez avec un œil nouveau vos patients avec une neuropathie sensitive non étiquetée et interrogez-les précisément sur la présence d’une toux, de signes vestibulaires et vérifiez leur IRM. Il s’agit d’une pathologie récessive, les cas apparemment sporadiques sont donc fréquents. Le diagnostic en biologie moléculaire du syndrome CANVAS se met actuellement en place en France.
D’après la communication d’Andrea Cortese (Londres). CANVAS: discovery of the gene for a common sensory ataxic neuropathy. Session 1, CANVAS/Sensory neuropathy. 24e édition des Journées francophones du nerf périphérique, 7 février 2020.
L’imagerie nerveuse est-elle l’avenir de l’électroneuromyogramme ?
L’imagerie neurologique a, de longue date, conquis le cœur des neurologues spécialistes des pathologies du système nerveux central et a révolutionné le diagnostic et le suivi de ces pathologies. Côté système nerveux périphérique, va-t-elle maintenant remplacer notre bon vieil électroneuromyogramme (ENMG) ?
L’échographie nerveuse
Les Drs Puma et Bouhour ont précisé la place grandissante de l’échographie nerveuse dans le bilan des neuropathies périphériques, et notamment des neuropathies dysimmunitaires. De nombreuses publications montrent ainsi l’intérêt croissant de l’échographie nerveuse dans le diagnostic des polyradiculonévrites chroniques (PRNC), des neuropa thies à anticorps anti-MAG, avec la mise en évidence d’élargissements multifocaux du diamètre des nerfs périphériques, ou même dans le syndrome de Parsonage Turner, avec visualisation de phénomènes de torsion du nerf.
- La sensibilité de l’échographie pour détecter une PRNC est de 97,4 %, avec une spécificité de 69,4 %.
- L’ENMG a quant à lui une sensibilité de 78,9 %, avec une spécificité de 93,5 %.
- La valeur ajoutée de l’échographie est de 21,1 % par rapport à l’étude ENMG seule pour le diagnostic de PRNC.
L’échographie nerveuse et l’étude ENMG sont donc des techniques complémentaires avec une meilleure sensibilité de l’échographie, mais une meilleure spécificité de l’électromyogramme. La mise à disposition de sondes à ultra-haute fréquence devrait également améliorer la sensibilité de cet examen. En revanche, son intérêt dans le suivi des thérapeutiques des polyradiculonévrites inflammatoires démyélinisantes chroniques (PIDC) semble moins évident, puisque l’équipe du Dr Bouhour a montré une variation très décalée dans le temps des anomalies échographiques par rapport à la clinique ou à l’ENMG. Les anomalies échographiques commencent également à être décrites dans les vascularites ou la neuropathie sensitive de Wartenberg.
L’IRM
Le Dr Vandendries, radiologue, nous a également montré l’intérêt de l’IRM dans cette exploration nerveuse avec une résolution et une puissance d’image de plus en plus importantes. L’IRM trouve particulièrement sa place dans la recherche d’une atteinte nerveuse proximale ou en examen de débrouillage, préalablement à une échographie ciblée.
Il apparaît maintenant indispensable que les neurologues se forment à l’échographie nerveuse, technique complémentaire de l’ENMG – comme l’électrocardiogramme et l’échographie cardiaque sont devenus les outils indispensables aux cardiologues – au détriment de leur stéthoscope.
D’après les communications de :
Françoise Bouhour (Lyon) et Angela Puma (Nice). Intérêt de l’échographie dans le diagnostic positif et différentiel des neuropathies dysimmunes, comparaison des techniques d’imagerie (IRM versus échographie) dans le diagnostic des neuropathies dysimmunes, intérêt de l’échographie dans le suivi des neuropathies dysimmunes. Session 2 Controverse, « Échographie vs IRM du nerf » et pour quelles indications ? 24eédition des Journées francophones du nerf périphérique, 8 février 2020.
Christophe Vandendries (Paris). MR-neurography: what could you expect? Session 2 Controverse, « Échographie vs IRM du nerf » et pour quelles indications ? 24e édition des Journées francophones du nerf périphérique, 8 février 2020.
Quelle était la communication la plus originale ?
Le Dr Bouhassira nous a rappelé l’importance de la prise en compte de l’effet placebo dans les essais cliniques et notamment les essais cliniques sur la douleur. Il était notamment question d’un essai clinique comparant l’administration d’un antalgique par voie parentérale administré par une infirmière ou par un ordinateur avec une efficacité plus marquée pour les patients ayant eu une administration par l’infirmière !
La prise en compte de cet effet placebo dans le montage d’un essai randomisé contre placebo doit donc être soigneusement étudiée sous de multiples angles pour des critères subjectifs comme la douleur. Cet effet doit aussi nous inciter à rester modestes dans l’interprétation d’un essai clinique d’un traitement sans groupe placebo.
D’après la communication de Didier Bouhassira (Paris). Placebo effet. Session 4, Pain viewed from another perspective. 24e édition des Journées francophones du nerf périphérique, 7 février 2020.
L’auteur déclare ne pas avoir de lien d’intérêt.
Correspondance
armelle.magot@chu-nantes.fr
« Checkpoint inhibitors » et neuropathies périphériques
par Cécile Cauquil
Quelles sont les complications immunologiques en lien avec l’immunothérapie anti-cancéreuse ?
L’immunothérapie anti-cancéreuse repose sur l’induction d’une réaction du système immunitaire contre les cellules tumorales, en inhibant les points de contrôle immunitaire : interaction entre une protéine de surface CTLA-4 (Cytotoxic T-lymphocyte Associated Protein 4) ou PD-1 (Programmed cell Death protein 1) sur les lymphocytes T et son ligand PDL-1 (Programmed Death Ligand protein 1). Les inhibiteurs des checkpoint (ICP) lèvent cette tolérance immunitaire. La classe thérapeutique comporte les anti-CTLA-4, anti-PD-1 et anti-PDL-1. L’efficacité de ces molécules a été démontrée au cours d’essais de phase III dans le mélanome métastatique, le carcinome épidermoïde pulmonaire, le cancer rénal et les tumeurs urothéliales, avec un bénéfice net sur la survie et une réponse prolongée dans le temps. Les indications en hématologie et oncologie sont croissantes, de nombreux essais thérapeutiques avec ces molécules sont en cours.
Cette levée d’inhibition est à l’origine de la survenue d’effets indésirables (EI) en lien avec l’induction d’une auto-immunité. Les EI immu nologiques peuvent affecter divers organes, les plus fréquents étant les atteintes cutanées, digestives (colites ou pathologie inflammatoire digestive) et endocriniennes. Ces effets restent peu fréquents et sont d’intensité modérée selon la classification CTCAE (Common Terminology Criteria for Adverse Events) grade de 1 à 4. Les atteintes du système nerveux central et périphérique restent exceptionnelles.
Quelles sont les atteintes du système nerveux périphérique ?
La fréquence des atteintes neurologiques est estimée entre 2,4 et 4,9 % selon les séries, de 1 à 3,8 %, pour les anti-CTLA-4, de 2,5 à 6, 1 % pour les anti-PD-1/PDL-1 et jusqu’à 14 % en cas de thérapie combinée. Elles sont le plus souvent peu sévères (grade 1 ou 2 CTCAE).
Les EI d’origine immunologique affectant le système nerveux périphérique sont :
- les polyradiculonévrites ou pseudo Guillain-Barré,
- les radiculopathies,
- les atteintes musculaires type myosite,
- les syndromes myasthéniques (possible forme frontière entre ces deux derniers)
- et enfin les atteintes isolées d’une paire crânienne qui peuvent s’intégrer dans les atteintes suscitées (myasthénie et PRN) ou dans le cadre d’une atteinte centrale.
La coexistence de différentes toxicités a été rapportée avec l’association d’une atteinte centrale et périphérique et la présence de manifestations extra-neurologiques. La survenue est le plus souvent précoce, dans les premiers mois suivant l’introduction du traitement (délai médian de 45 jours), mais avec des intervalles variant de 1 à plus de 170 jours.
Quelle est la prise en charge des effets indésirables immunologiques ?
Des recommandations ont été rédigées par les sociétés européenne et américaine sur la gestion des toxicités des ICP, proposant des algorithmes pour le diagnostic, la présentation clinique, les explorations complémentaires et la prise en charge thérapeutique. Une fois le diagnostic suspecté, la prise en charge repose sur l’arrêt de l’immunothérapie, l’instauration d’une corticothérapie dont la dose et la voie d’administration seront adaptées à la sévérité de l’EI. On recommande généralement 1 mg/ kg/ jour d’équivalent prednisone per os pour les EI de grade ≥ 2, qui pourra être augmentée à 2 mg/kg/ jour ou administrée sous forme de bolus IV pour les situations graves. La corticothérapie est à décroître sur une période de 4 à 6 semaines pour éviter un effet rebond en lien avec la demi-vie prolongée des ICP. La corticothérapie se révèle efficace, y compris dans les situations cliniques habituellement réfractaires, tel le syndrome de Guillain-Barré. Le traitement et les mesures symptomatiques seront à adapter en fonction de l’évolution clinique.
La reprise de l’ICP se discutera au cas par cas en fonction de la sévérité de l’EI, de la récupération et de la réponse tumorale.
Quelle attitude adopter dans les situations réfractaires ?
Parmi les EI immunologiques neu-rologiques, certains se montrent réfractaires au traitement par corti-coïdes seuls. Les échanges plasmatiques ou les IgIV sont fréquemment utilisés en deuxième ligne ou d’emblée dans certaines situations à risque ou sévères (grade ≥ 3) telles qu’un syndrome myasthénique, une encéphalite ou une polyneuropathie inflammatoire. Des immunosuppresseurs ciblant des phases spécifiques de l’inflammation aiguë pourraient être des pistes pour le traitement des EI réfractaires comme cela est réalisé en pratique pour les colites traitées par anti-TNFaα. Des molécules telles que les inhibiteurs de l’IL-6 ou de l’IL-10, ou les anti-CD20, ont été proposées dans certaines situations.
La prise en charge de ces situations complexes nécessite une bonne collaboration entre les oncologues, immunologistes et neurologues, idéalement dans le cadre d’un réseau ; ceci afin d’optimiser la prise en charge et prendre les décisions de traitement en concertation.
How to manage neurological complications of ICI and CIDP associated with cancer. Session 2, Immune mediated neuropathies associated with cancer. 24e édition des Journées francophones du nerf périphérique, 7 février 2020.
L’auteur déclare ne pas avoir de lien d’intérêt.
Correspondance
cecile.cauquil@aphp.fr
Bibliographie
– Brahmer JR, Lacchetti C, Schneider BJ et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline. J Clin Oncol 2018 ; 36 : 1714–68.
– Anderson D, Beecher G, Nathoo N et al. Proposed diagnostic and treatment paradigm for high-grade neurological complications of immune checkpoint inhibitors. Neurooncol Prac 2019 ; 6 : 340–45.
– Martins F, Sykiotis GP, Maillard M et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol 2019 ; 20 : e54-e64.
Domaine métabolique, quelles sont les nouveautés ?
par Lucie de Clerck
Dans l’obésité
L’obésité est un facteur de risque car diovasculaire connu avec de nombreuses comorbidités associées, notamment le diabète de type 2. Il a été montré une association entre obésité et développement de neuropathie périphérique, de manière indépendante à la présence ou non d’un diabète [1]. Certains biomarqueurs pro-inflammatoires retrouvés au niveau du tissu adipeux pourraient jouer un rôle dans cette association [1]. La prise en charge de l’obésité est donc primordiale. Il a ainsi été mis en évidence que la chirurgie bariatrique diminuait le risque de complications microvasculaires ainsi que celui de développer une neuropathie.
Dans le diabète de type 2
Le diabète de type 2 est également un facteur de risque connu de développer une neuropathie, c’est même l’une des causes les plus fréquentes de neuropathie en France. Son contrôle, via la surveillance de l’HBA1c, est l’un des objectifs dans la prise en charge des neuropathies diabétiques. Il a été montré que le contrôle intensif du diabète de type 2, avec un objectif de normalisation de l’HBA1c, permettait une amélioration – pas seulement une stabilisation – des neuropathies diabétiques [2].
La metformine
La metformine est l’un des antidiabétiques oraux les plus prescrits. Sa prise peut entraîner une carence en vitamine B12 avec une relation entre la dose et la durée du traitement. La question concernant le risque de développer une neuropathie en lien avec cette carence vitaminique a été soulevée. Cependant, de manière rassurante, il n’a pas été montré de sur-risque de développer une neuropathie lors de la prise de metformine [3].
Les statines
Enfin, l’association entre neuropathie et traitement par statine fait également l’objet de débats. Certains cas de neuropathie survenant sous statine et régressant après arrêt du traitement hypolipémiant ont été décrits. Cependant, peu d’études ont été réalisées jusqu’à présent. Une étude récente s’est penchée sur la question et a montré des données rassurantes avec l’absence de sur-risque de neuropathie en cas d’exposition aux statines [4]. Il restera à faire attention au muscle.
L’auteur déclare ne pas avoir de lien d’intérêt.
Correspondance
lucie.declerck@chu-nantes.fr
Bibliographie
2. Ishibashi F, Taniguchi M, Kosaka A et al. Improvement in neuropathy outcomes with normalizing HbA1c in patients with type 2 diabetes. Diabetes Care 2019 ; 42 : 110-8.
3. Yang W, Cai X, Wu H, Ji L. Associations between metformin use and vitamin B12 levels, anemia, and neuropathy in patients with diabetes: a meta-analysis. J Diabetes 2019 ; 11 : 729-43.
4. Warendorf JK, Vrancken AFJE, van Eijk RPA et al. Statins do not increase risk of polyneuropathy: A case-control study and literature review. Neurology 2019 ; 92 : e2136-e2144.
Zoom sur quelques posters : un peu de génétique, un peu de physiopathologie !
par Yann Péréon
Mutation sur le gène MORC2 et amyotrophie spinale
La très grande majorité des amyotrophies spinales est liée à des mutations sur le gène SMN1, mais plus d’une vingtaine d’autres gènes peuvent également en être responsables. Les auteurs ont identifié une mutation hétérozygote sur le gène MORC2 chez une femme de 43 ans qui présentait un tableau associant crampes, déficit distal aux membres supérieurs, proximal aux membres inférieurs, avec un EMG neurogène dans les mêmes territoires. Cette mutation, jusque-là jamais rapportée, affecte le domaine S5 du gène connu pour présenter des mutations pathogènes. Cette première identification de mutation de MORC2 dans un cas d’amyotrophie spinale permet d’élargir le spectre des phénotypes cliniques associés à ce gène par ailleurs responsable de CMT2Z [2].
Masingue M, Latour P, Stojkovic T. Spinal muscular atrophy cause by an original MORC2 mutation. Poster NH.06. 24e édition des Journées francophones du nerf périphérique.
Mutations sur le gène LRSAM1 et neuropathie
Les mutations affectant le gène LRSAM1 sont responsables de formes axonales de Charcot-Marie-Tooth (CMT), dites CMT2P, soit dominantes, soit récessives. Le phénotype habituellement rapporté est celui de formes tardives et à prédominance sensitive, avec pieds creux [1]. Une élévation associée des CPK n’est pas exceptionnelle. Gunawardana et al. rapportent deux nouveaux cas sporadiques chez des patientes non apparentées, avec un début dans la trentaine.
Le début sensitif était notable : l’une se faisait pénaliser au netball (un sport dérivé du basket-ball) pour marcher sur les pieds des autres joueuses sans s’en rendre compte, l’autre présentait régulièrement des ampoules aux pieds qui restaient indolores.
Le tableau clinico- électrophysiologique était celui d’une neuropathie axonale, longueur dépendante, à prédominance sensitive nette, mais avec une composante motrice en détection dans les muscles des membres inférieurs. Des mutations hétérozygotes ont été mises en évidence dans ces deux cas, dont l’une jusqu’ici non rapportée. LRSAM1 code pour une ubiquitine-ligase de type E3, exprimée dans les motoneurones spinaux et les neurones sensitifs, jouant un rôle dans le développement et le fonctionnement des nerfs. Ce gène, connu depuis une dizaine d’années pour être lié à des neuropathies, fait maintenant partie des panels spécifiques testés à large échelle en France.
Gunawardana N, Roberts R, Reid E (Cambridge). Mutations in LRSAM1 causing a mild sensory predominant neuropathy. Poster NH.01.24e édition des Journées francophones du nerf périphérique, 7 février 2020.
Anticorps anti-FGFR3 et neuropathies
Il n’est pas toujours facile d’identifier la cause d’une neuropathie sensitive, celle-ci reste bien souvent qualifiée d’idiopathique. Une origine dysimmunitaire est parfois suspectée, et même retrouvée, et l’équipe de Saint-Étienne a ainsi démontré l’association neuropathie sensitive/anticorps anti-FGFR3 (Fibroblast Growth Factor Receptor 3) [3]. La question reste ensuite posée sur des liens exacts entre les deux (ex. : causalité ou pas ?).
Pour avancer dans ce sens, nos collègues de la Loire se sont intéressés, d’une part, aux cibles précises des anticorps, d’autre part, aux voies de signalisation en aval du récepteur FGFR3 (Fig.1). Pour les premières, ils ont réussi à les identifier dans un quart de leur population de patients (15/66), il pouvait dans certains cas s’agir du domaine tyrosine kinase, critique dans le lancement de la voie de signalisation cellulaire ; ces patients présentaient un phénotype clinique et ENMG plus sévère, élément en faveur d’un lien de pathogénicité de ces anticorps. Pour les secondes, les auteurs ont travaillé sur des neurones corticaux de souris soumis à des anticorps anti-FGFR3 de lapins ou de patients porteurs de neuropathie sensitive et anticorps. Parmi les effets observés, une augmentation de la mort cellulaire, une hyperexpression de récepteurs NMDA au glutamate, ou encore l’activation de processus d’autophagie.
Ça avance et c’est un bel exemple de progrès à venir, sans doute un peu similaires à ceux qu’on connaît pour d’autres neuropathies dysimmunitaires avec anticorps anti-neurofas cine et autre anti-Caspr.
Nasser Y, Reynaud-Federspiel E, Camdessanché JP et al. The role and molecular mechanisms of the anti-FGFR3 antibody, a biomarker of sensory neuropathies inducing neuronal death. Poster NS.05. 24e édition des Journées francophones du nerf périphérique.
Tholance Y, Antoine JC, Mohr L et al. Anti-FGFR3 antibody epitopes are functional sites and correlate with the neuropathy pattern. Oral posters presentations. 24e édition des Journées francophones du nerf périphérique, 7 février 2020.
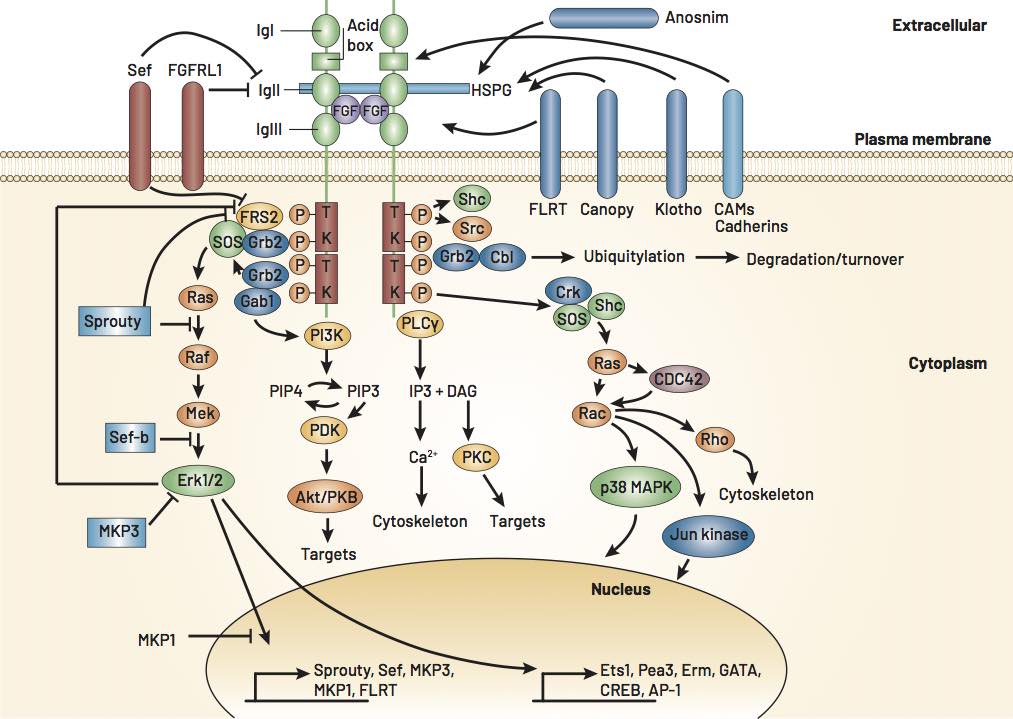
Figure 1 – Voies de signalisation des récepteurs aux facteurs de croissance des fibroblastes FGFR (D’après Mason I. Nat Rev Neurosci 2007 ; 8 : 583-96).
Chaînes légères des neurofilaments (NfL) et transthyrétine
Les avancées thérapeutiques de ces dernières années ont transformé la prise en charge des neuropathies amyloïdes à transthyrétine [4]. Cette protéine, principalement produite par le foie, peut, lorsqu’elle est mal constituée, s’accumuler et former des dépôts au sein de nombreux organes, principalement mais pas uniquement les nerfs et le cœur.
Les traitements actuels permettent d’en réduire la synthèse hépatique ou de stabiliser la protéine et d’éviter son accumulation anormale. Leurs effets sont évalués sur des scores cliniques (et c’est tant mieux, l’objectif étant d’améliorer les patients, pas les marqueurs), mais le besoin en biomarqueurs objectifs reste critique, dans ce domaine comme dans de nombreux autres en pathologie neuromusculaire (ex. : la SLA). Les auteurs, partant des données de l’essai thérapeutique fondé sur des ARN interférents réduisant l’expression de la transthyrétine, se sont intéressés aux taux plasmatiques de NfL. Les chaînes légères des neurofilaments, composants structurels essentiels des fibres nerveuses, représentent un marqueur de lésion neuro-axonale aussi bien dans le système nerveux central que périphérique. Ici, les taux sériques de NfL, symboles de dommages nerveux, étaient corrélés avec les scores cliniques des patients (mNIS+7, utilisé dans les neuropathies hATTR) : d’autant plus bas que l’amélioration induite par le traitement était élevée. Les NfL représentent probablement un outil bien utile dans le futur pour le suivi des neuropathies, sans doute pas uniquement hATTR.
Ticau S, Sridharan G, Tsour S et al. Neurofilament light chain (NfL) as a potential biomarker in hereditary transthyretin-mediated (hATTR) amyloidosis. NH.05. 24e édition des Journées francophones du nerf périphérique.
L’auteur déclare ne pas avoir de lien d’intérêt.
Correspondance
Yann.Pereon@univ-nantes.fr
Bibliographie
2. Sevilla T, Lupo V, Martínez-Rubio D et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain 2016 ; 139 : 62-72.
3. Tholance Y, Moritz CP, Rosier C et al. Clinical characterisation of sensory neuropathy with anti-FGFR3 autoantibodies. J Neurol Neurosurg Psychiatry 2020 ; 91 : 49-57.
4. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019 ; 15 : 387-404.